

Synergistic and Adaptive Organometallic Chemistry
Our research explores the concept of synergistic organometallic chemistry, where multiple entities cooperate in non-trivial ways to activate and transform chemical bonds. Unlike classical approaches that rely on the linear and isolated action of a single reagent or catalyst, these systems exploit dynamic and interconnected interactions, giving rise to emergent properties and adaptive modulation of reactivity.
Inspired by the flexibility of natural enzymatic systems, this approach enables the design of molecular entities capable of adjusting their reactivity and selectivity in response to their local environment (ligand architecture, solvent polarity, pH, redox potential) or external stimuli (light, electric current). Such strategies open promising perspectives for energy conversion and storage, where fluctuating energy inputs and variable feedstock quality demand adaptive, robust, and universal catalytic solutions.
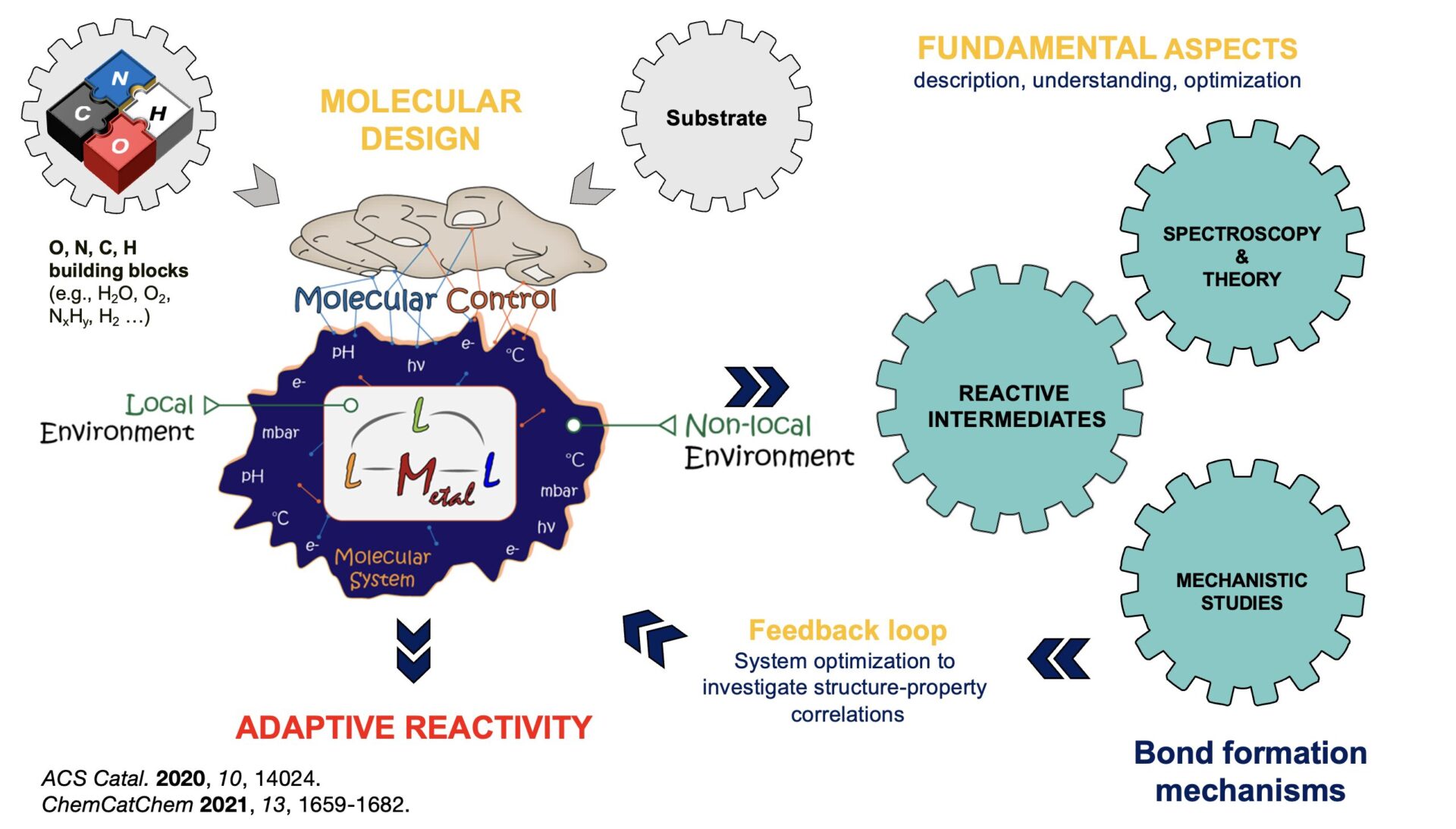
Molecular approach to the development of dynamic and adaptive systems
Synergistic Organometallic Chemistry and Heterobimetallic Systems
Our research also focuses on the development of molecular transformations through synergistic organometallic pathways and the mechanistic understanding of these processes. We specifically investigate the role of heterobimetallic species, in which two metal centers cooperate in concert to modulate reactivity and selectivity. This approach provides access to regio-, chemo-, stereo-, and even divergent behaviors that remain unattainable with classical monometallic species.
A representative example is the cooperation between lithium and zinc: the combination of a highly reactive but poorly selective organolithium reagent with a selective but modestly reactive organozinc compound produces a mixed species with enhanced reactivity and selectivity. This type of synergistic interplay illustrates the “PAIRiodic Table” concept, where unique heterobimetallic entities are envisioned as new chemical elements in their own right.
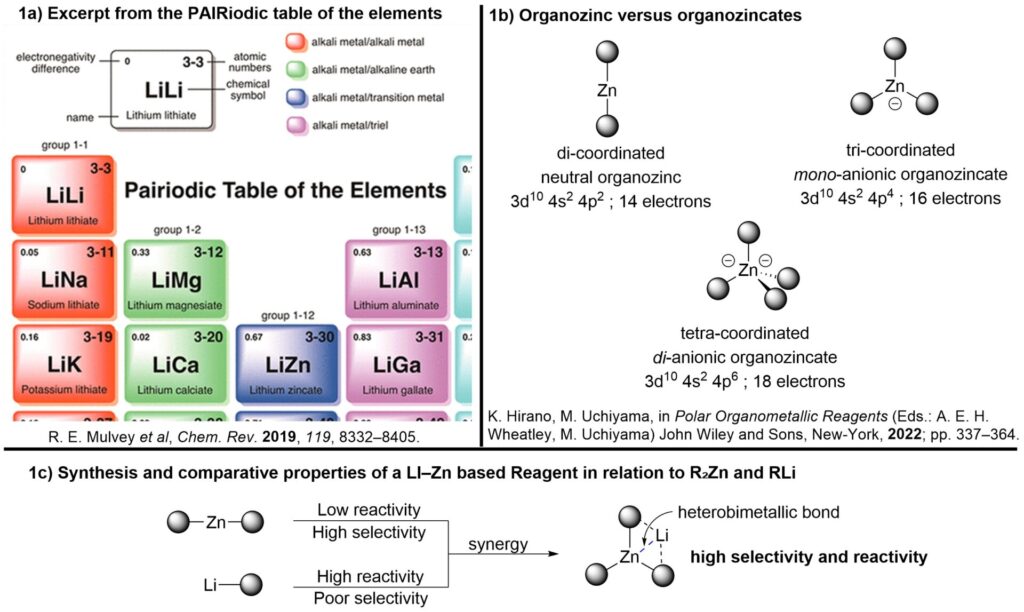
Excerpt from the PAIRiodic table of elements with a zoom on the LiZn element
Divergent Bond Activation
One of our projects explored whether a catalyst could open divergent reaction pathways based on reaction conditions by selectively targeting the intrinsic chemical bonds of a primary functional group.[1] Using aldehydes as a model substrate, we developed an iron-based catalyst system capable of selectively addressing C=O (Figure blue) and C-H (Figure, red) to produce nitriles or amides under mild conditions. Mechanistic studies revealed that different active species, such as neutral Fe(III)Br3 or an ion pair [Fe(III)Br4][N(C4H9)4], drive these transformations.
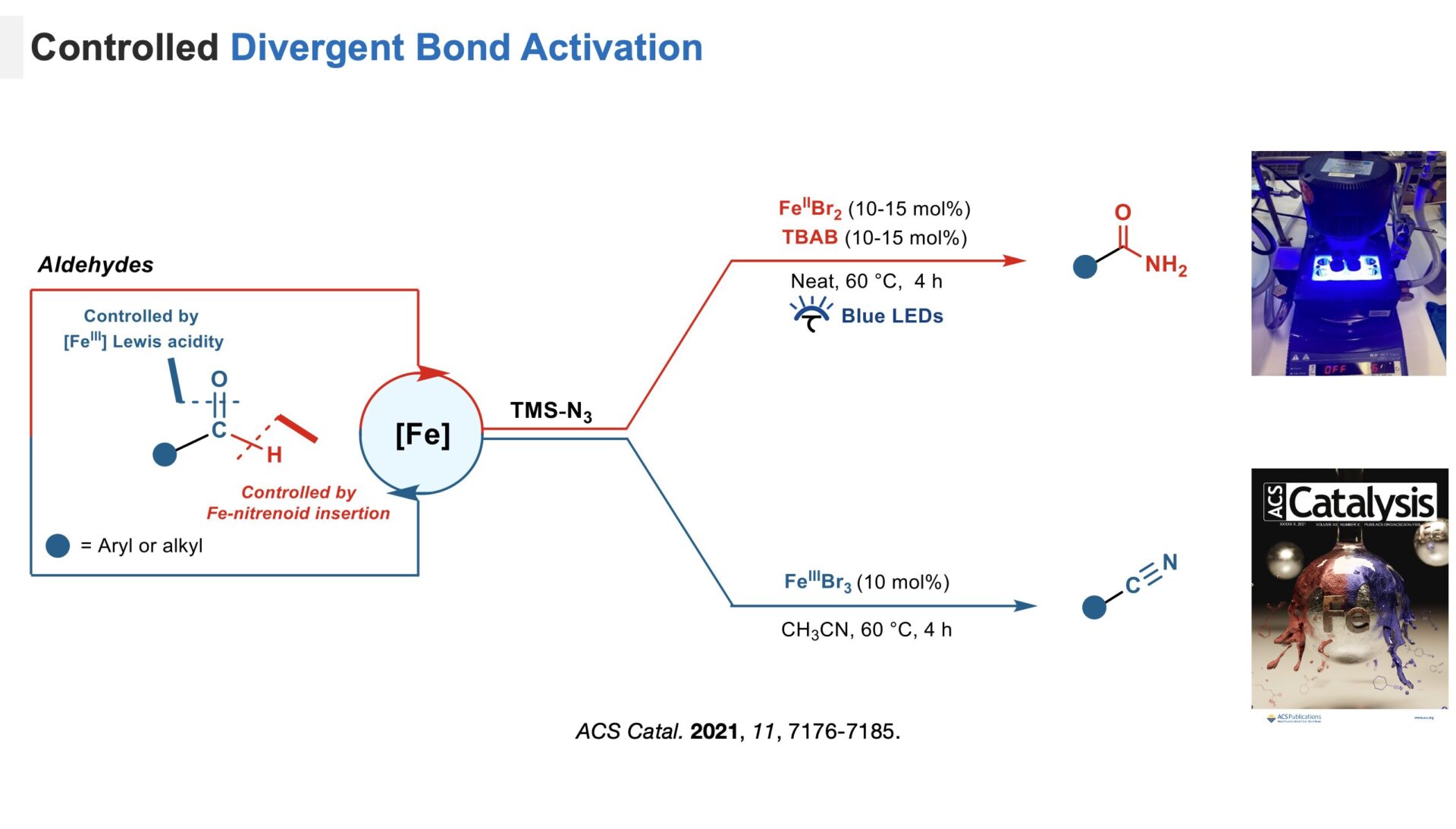
Development of an Iron-based system for divergent bond activation
Adaptive Catalytic Systems
To develop adaptive platforms, we designed a catalytic system featuring a ligand with a borane arm to create a polarized environment that interacts with a rhodium metal center.[2] This system, tested for the hydrogenation of nitroarenes, demonstrated selective control over reduction levels, producing either fully reduced anilines or hydroxylamines under mild conditions. This approach highlights the potential of adaptive catalysts to manage complex reaction networks selectively.
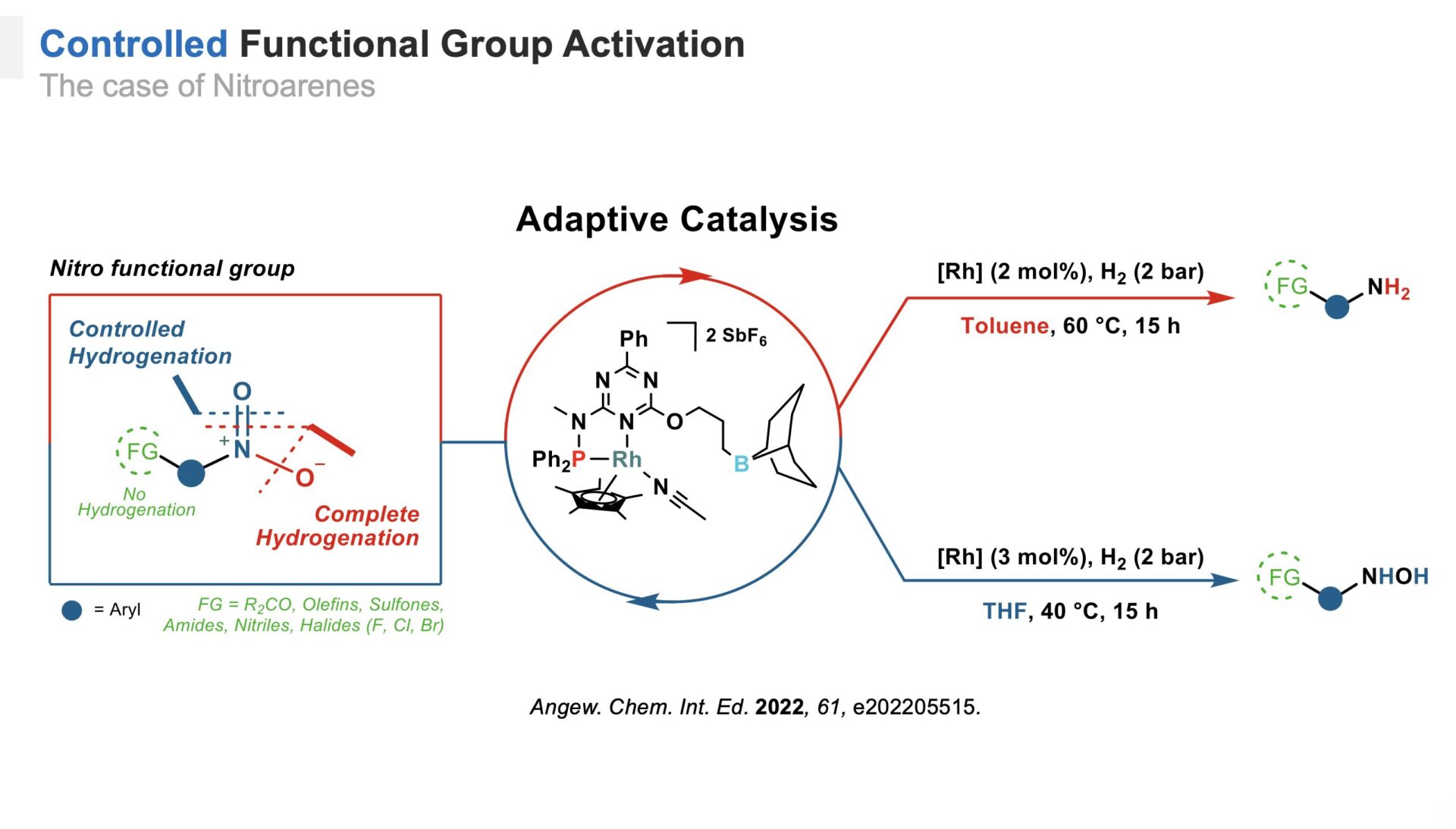
Adaptive hydrogenation of nitroarenes.
One-Pot Dual C–C Bond Formation
We found that 2-MeTHF promotes the zincate-mediated remote functionalisation of p-iodobenzyl derivatives through metallotropy with a broad scope.[3] 2 C-C bonds are formed at sites distant by 5 atoms in a single operation and by using only one reagent. The biosourced solvent remarkably impacted all the key elementary steps involved in the tandem reaction. The method tolerates a wide range of sensitive functionalities within the substrates as well as enolisable and activated or deactivated aromatic aldehydes as reaction partners. A benzyl naphthalene derivative was even reacted through an original dearomatisation / rearomatisation / metallotropy chain.
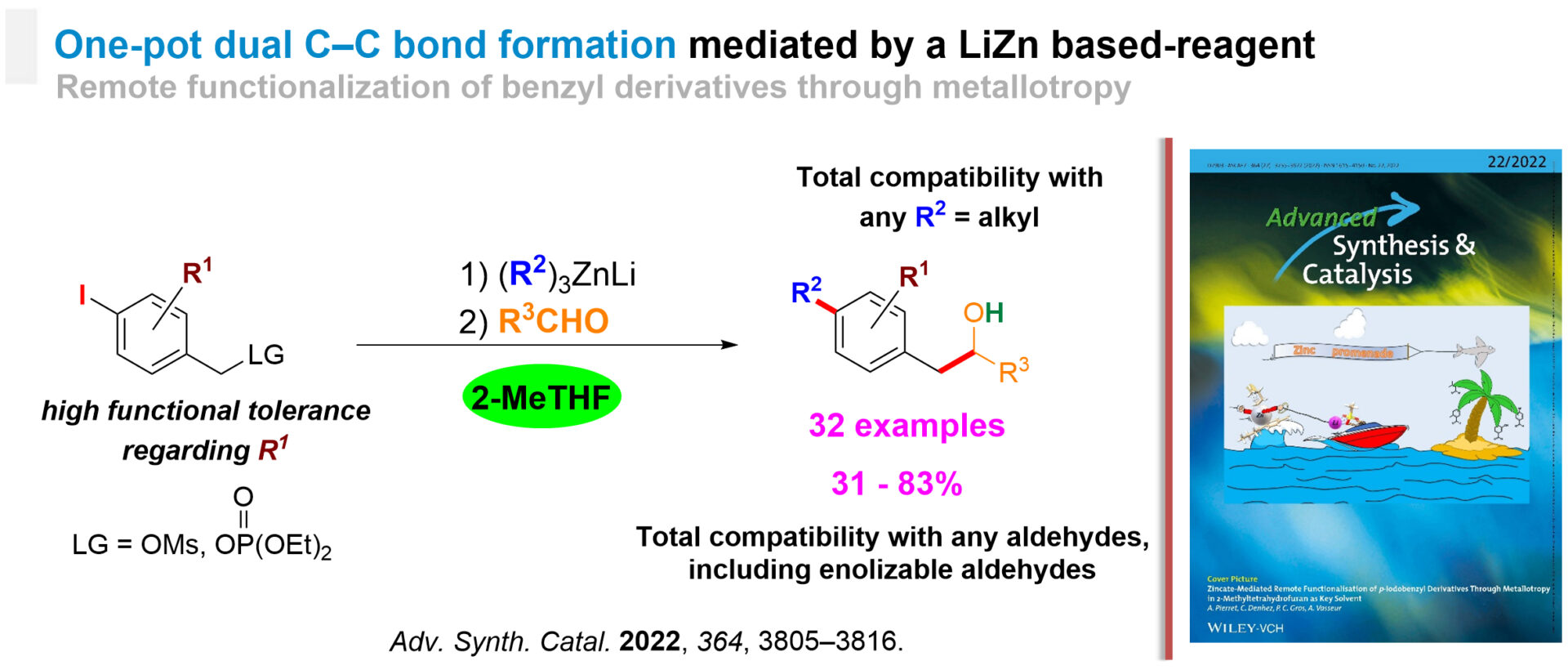
A broad-spectrum method for the regioselective dual C-C bond formation in benzyl derivatives.
Enhanced Bond Activation
Addressing the challenge of selective C–F bond activation, we developed a cooperative system combining a Rh(DMPE)2H fragment with secondary phosphine oxide.[4] This system catalyzes the hydrodefluorination of perfluoroarenes with high chemoselectivity. Aside from substrates with electron-withdrawing functional groups, the system showed an exceedingly rare tolerance for electron-donating functionalities and heterocycles. Mechanistic insights revealed a rhodium(I) dihydride and phosphine oxide as key cooperative elements, emphasizing the value of molecular templates in overcoming challenging bond activations.
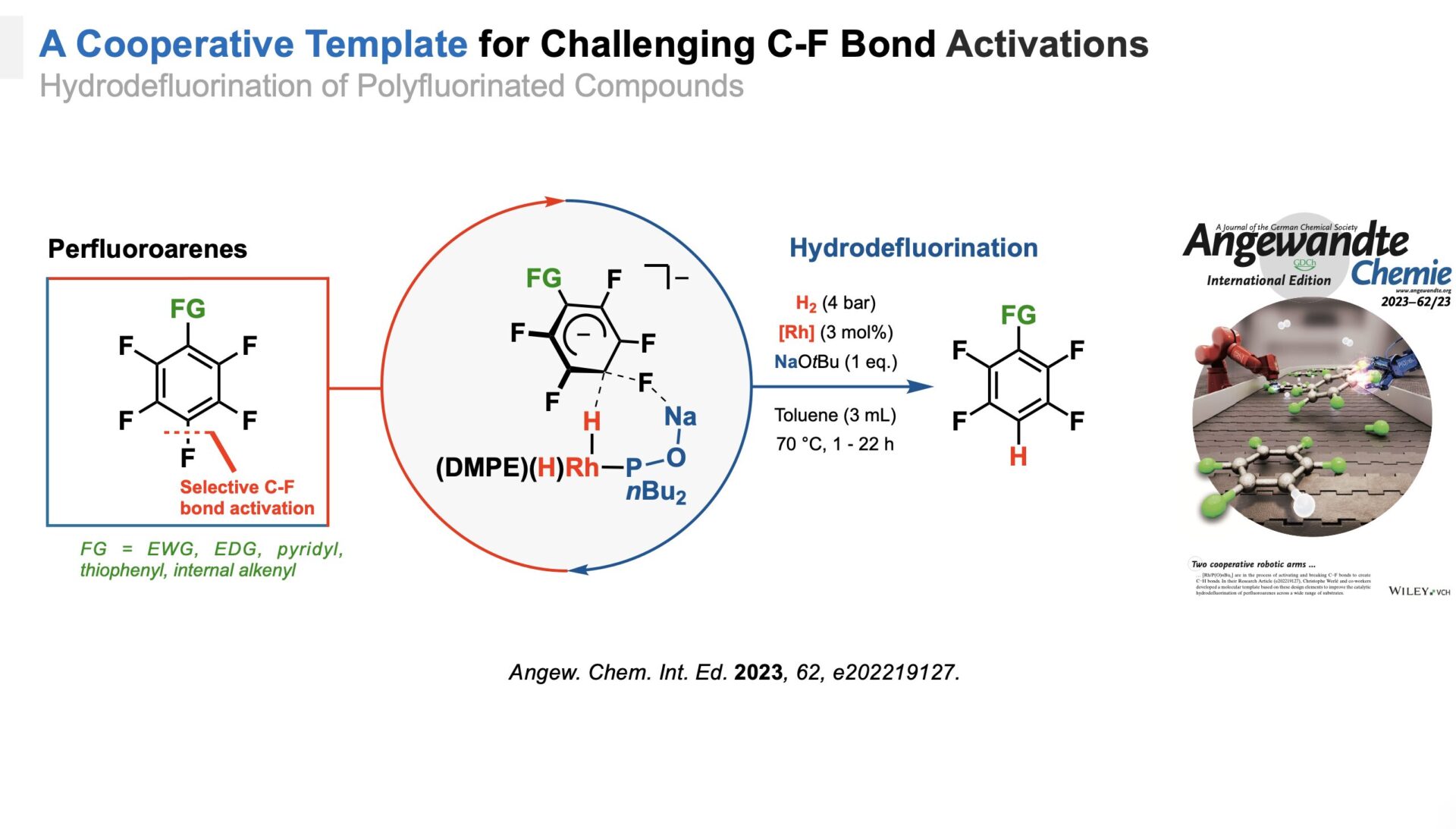
A cooperative template for enhanced C-F bond activation.
Carbon-Carbon Bond Formation
Focusing on carbon-carbon bond formation, we designed a cobalt-boron catalytic system that synergistically activates aldehydes and (trimethylsilyl)diazomethane.[5] This system efficiently facilitates the insertion of a single carbon atom into the substrate, producing silyl enol ethers under mild conditions while preserving sensitive functional groups. This dual-activation approach represents a significant advance in bond activation and molecular synthesis.
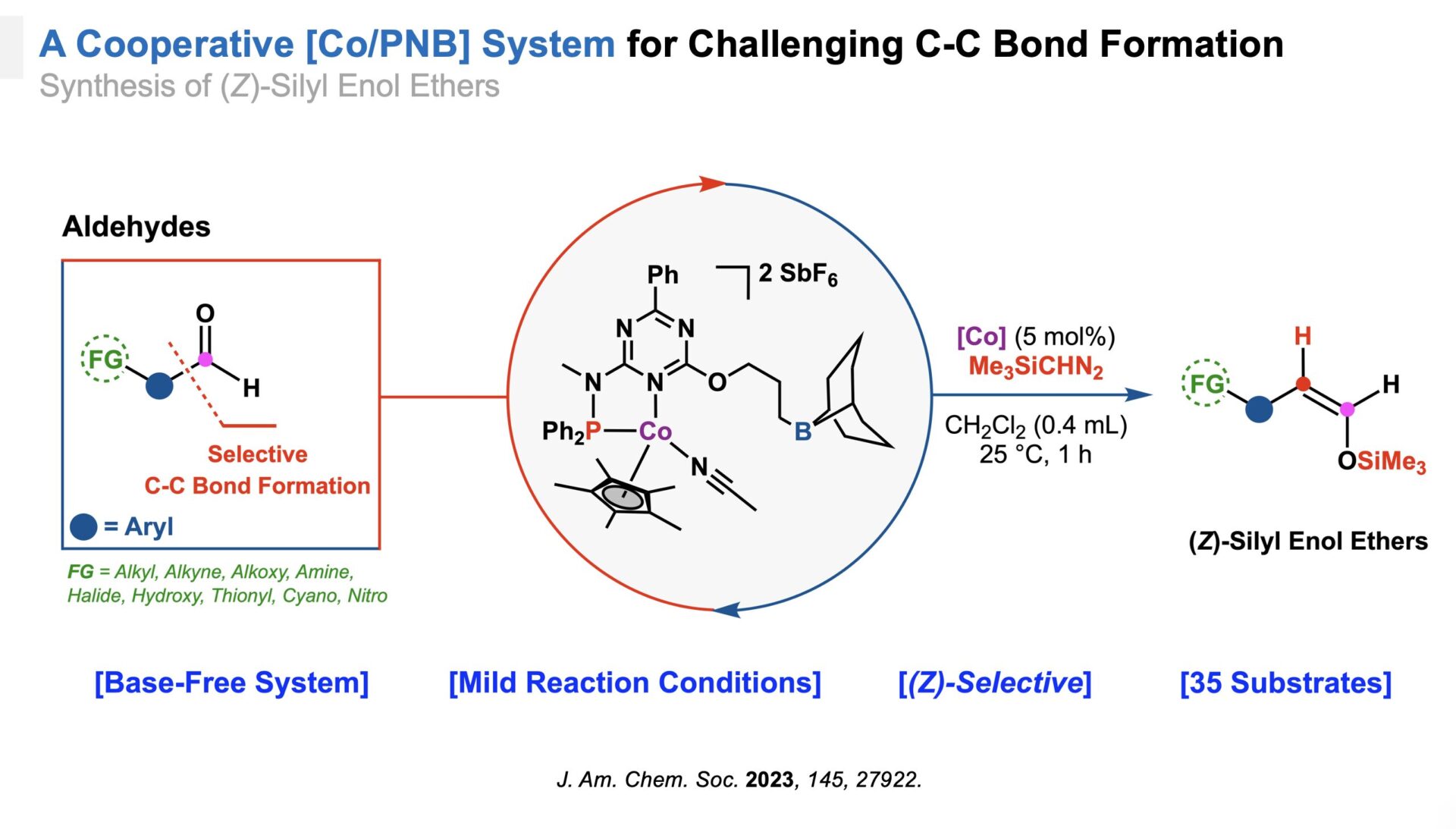
A cooperative [Co/PNB] System for C-C bond formation.
Synergistic Effect in a Li–Zn Complex
I/Zn Exchange reaction in THF has been revisited computationally through a microsolvation approach by considering Et3ZnLi·2THF as reagent and 4-iodobenzyl mesylate as substrate.[6] We found that Et3ZnLi·2THF exhibited 3 resonance forms in which a heterobimetallic bond could be computationally characterized for the first time. Based on these 3 resonance forms, 3 mechanistic pathways have been calculated. The pathway involving a THF-solvated open zincate complex turned out to be the most energetically favored. During the process, the heterobimetallic bond is weakened but maintained throughout the process, indicating that Zn and Li operate synergistically, as if they behaved as a single element, ‘LiZn’. Importantly, the I/Zn exchange was shown to proceed through a lithium-assisted aryl shuttle-like process. The iodoaryl substrate is first converted into ArLi intermediate which in turns reacts with the remaining diorganozinc reagent. This 2-step process echoes the so-called trans-metal-trapping mechanism.
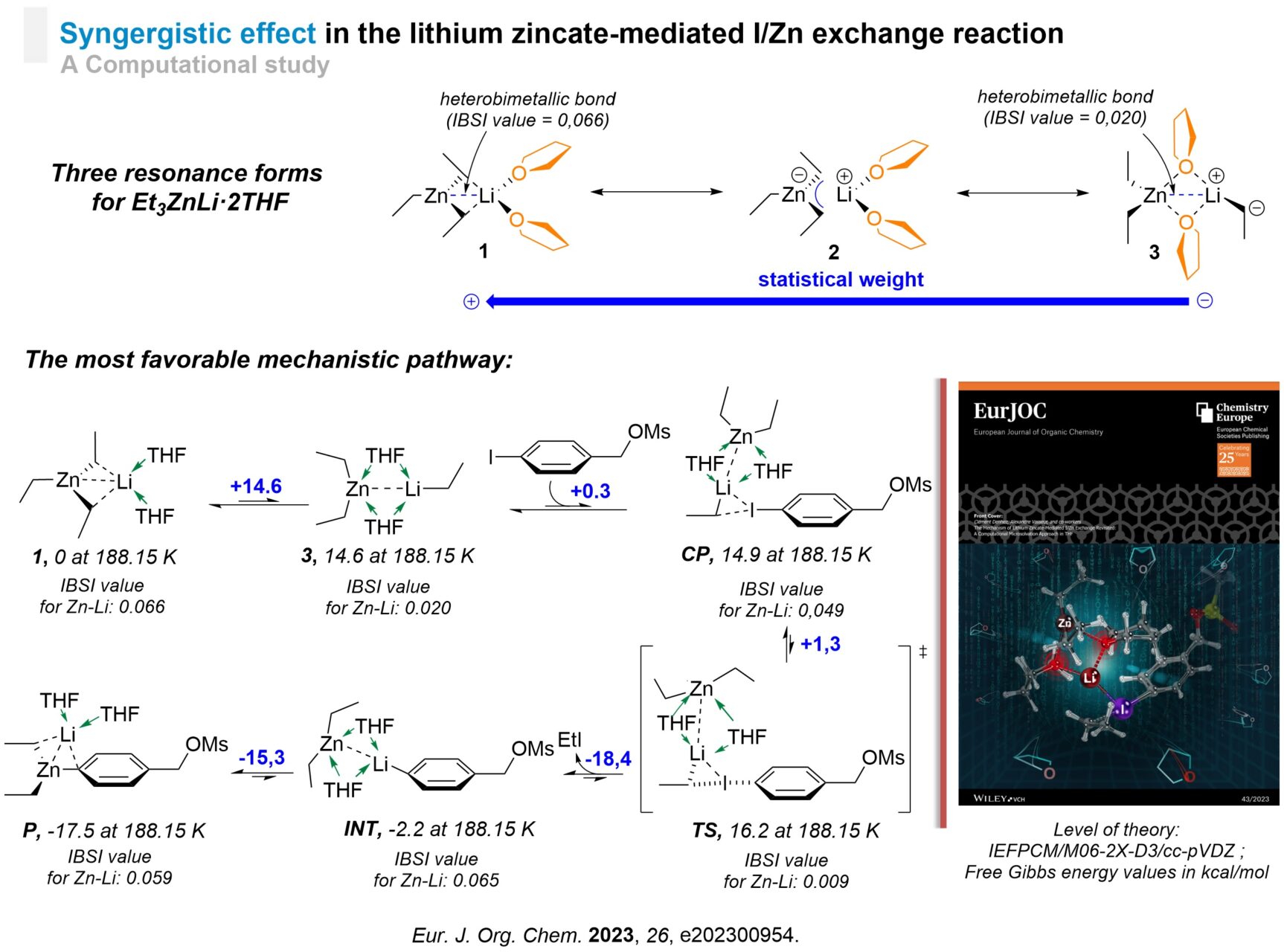
The Mechanism of Lithium Zincate-Mediated I/Zn Exchange Revisited.
Adaptive Alkyne Semihydrogenation
In our research on alkyne semihydrogenation, we demonstrate the transformative potential of adaptive catalysis to control not only chemo- and regioselectivity but also stereoselectivity.[7] We developed a catalyst with a uniquely engineered ligand environment to optimize hydrogen activation and transfer, enabling the selective synthesis of both cis- and trans-alkenes without additives. Mechanistic studies revealed the crucial role of the solvent in guiding unconventional hydrogenation pathways. Additionally, the catalyst’s exceptional durability over multiple cycles highlights its potential for advancing sustainable chemical processes.
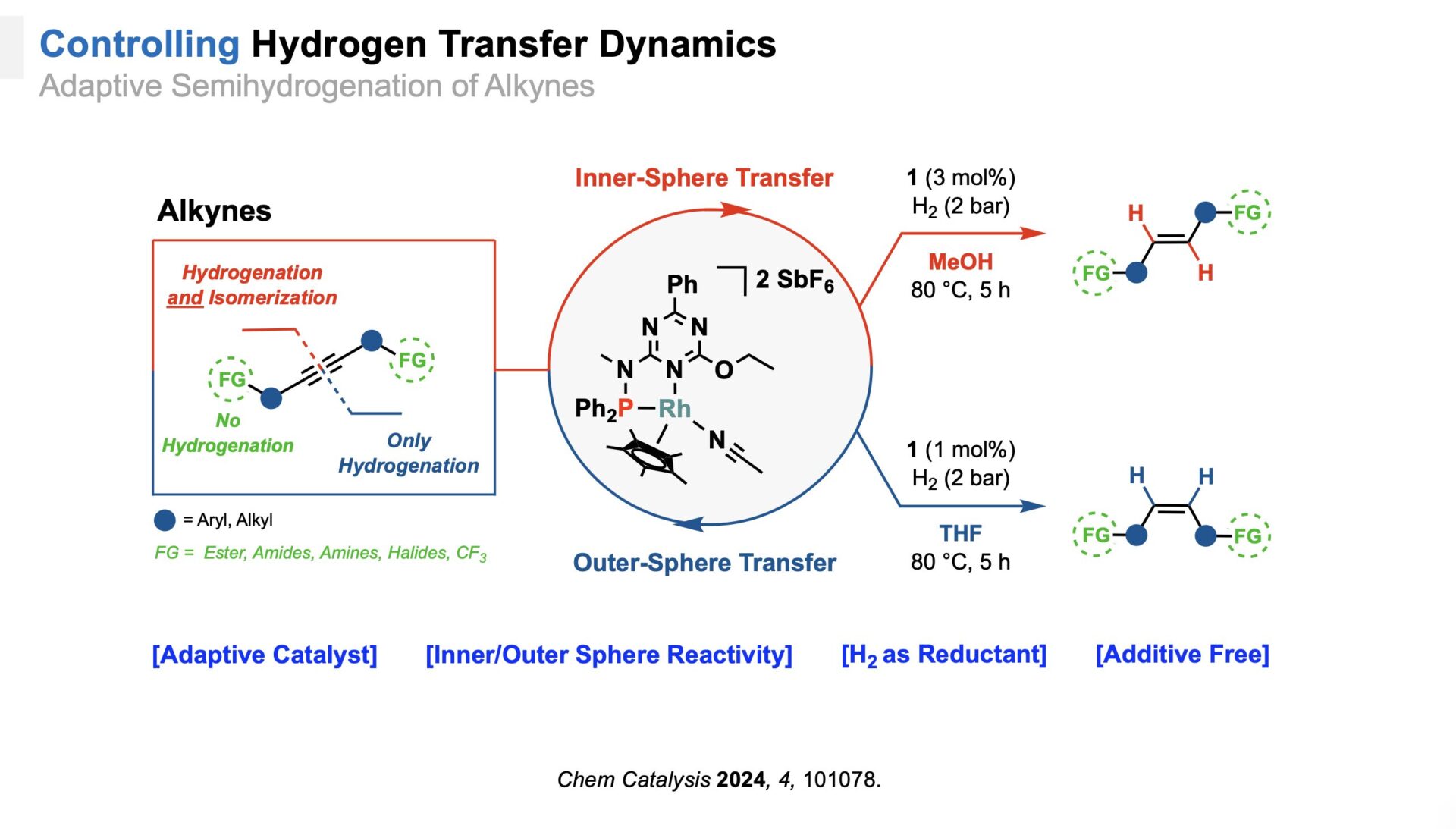
An adaptive Catalyst for alkyne semihydrogenation.
Divergent One-Pot Dual C–C Bond Formation
We developed a one-pot divergent synthesis promoted by a lithium organozincate enables the regioselective formation of two C–C bonds in thiophenes. [8] By simply modulating the zinc coordination environment, the same combination of reagents (ZnCl₂, an organolithium R¹Li, and a diethyl (5-halo)thienyl phosphate) affords two different products which are constitutional isomers—either through a sequence of consecutive reactions or via two independent intermolecular steps. This strategy allows for regioselective cross-couplings of the CAr(sp²)–Cthienyl(sp²) and Cthenyl(sp³)–CAr(sp²) types, without the need for transition metals or aryl partners pre-activated as boronic acids. The divergence is electron-count-controlled since the outcome of the transformation is only governed by the electron count at the zinc center of the involved organometallic reagent (either a 16-electron or an 18-electron species generated from ZnCl₂ and 3 or 4 equivalents of an organolithium reagent respectively).
With its broad synthetic applicability and reliance on earth-abundant, low-toxicity metals (Zn, Li), this method provides a sustainable approach for the synthesis of complex molecules with high potential in both pharmaceutical and materials sciences.
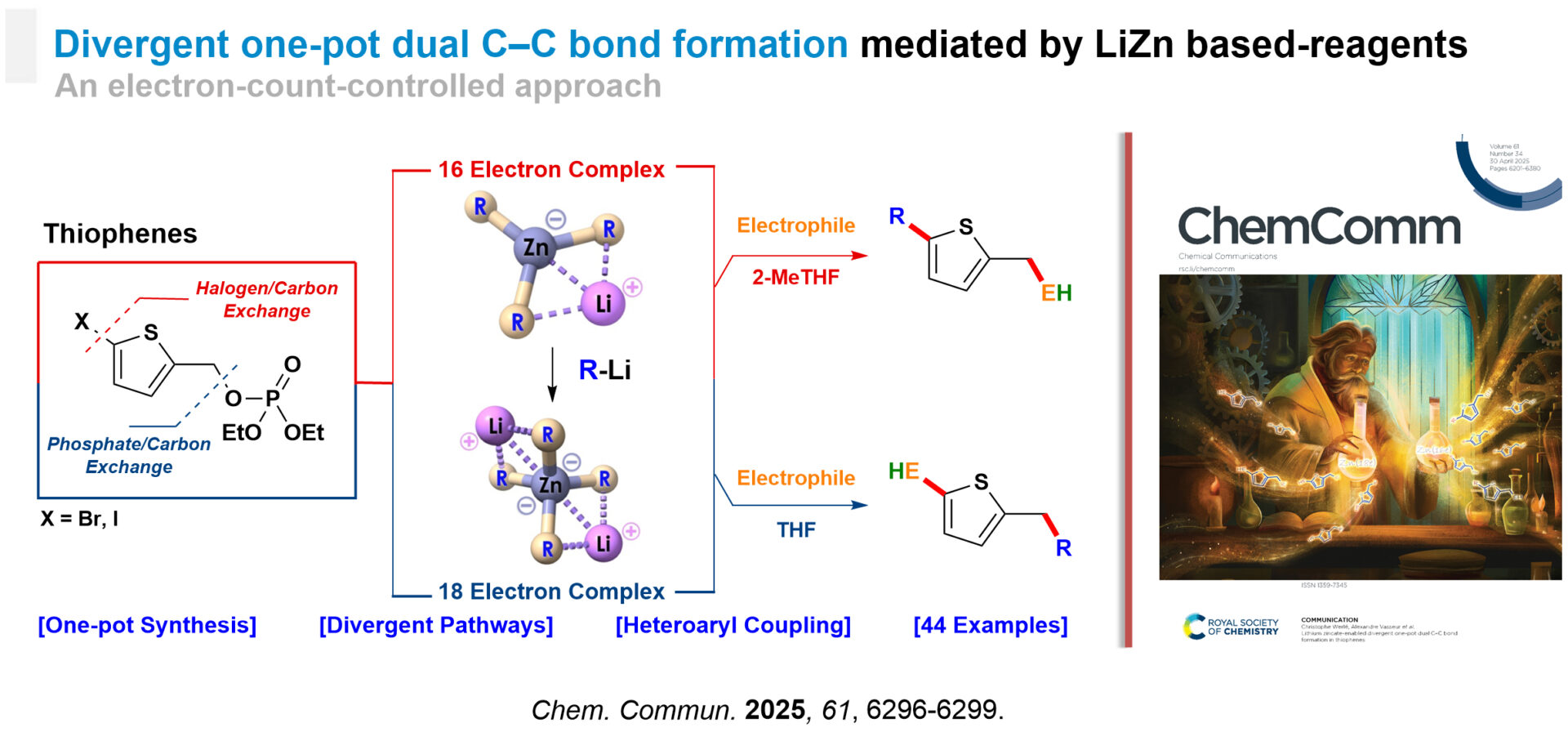
Electron-Count-Controlled Approach to Divergent Dual One-Pot C–C Bond Formation in Thiophenes.
Open Positions
The group of Synergistic Organometallic Catalysis (SynOCat) is always seeking new talented students. Exceptionally qualified applicants are welcome to get in touch with Dr. Werlé at any time (christophe.werle@univ-lorraine.fr). Such inquiries should include a curriculum vitae and a cover letter mentioning eligible fellowship funding agencies to whom you might apply to support your stay in the group. We are more than willing to assist you in the preparation of these applications. In addition, please have two letters of recommendation sent to Dr. Werlé by academic mentors who have previously supervised your work.
Bibliographie
[1] B. Chatterjee, S. Jena, V. Chugh, T. Weyhermüller, C. Werlé, ACS Catal. 2021, 11, 7176-7185.
[2] V. Chugh, B. Chatterjee, W. C. Chang, H. H. Cramer, C. Hindemith, H. Randel, T. Weyhermüller, C. Farès, C. Werlé, Angew. Chem., Int. Ed. 2022, 61, e202205515.
[3] A. Pierret, C. Denhez, P. C. Gros, A. Vasseur, Adv. Synth. Catal. 2022, 364, 3805–3816.
[4] W. C. Chang, H. Randel, T. Weyhermüller, A. A. Auer, C. Farès, C. Werlé, Angew. Chem., Int. Ed. 2023, 62, e202219127.
[5] S. Jena, L. Frenzen, V. Chugh, J. Wu, T. Weyhermüller, A. A. Auer, C. Werlé, J. Am. Chem. Soc. 2023, 145, 27922-27932.
[6] A. Pierret, C. Lefebvre, P. C. Gros, C. Denhez, A. Vasseur, Eur. J. Org. Chem. 2023, 26, e202300954.
[7] V. Chugh, J. Wu, M. Leutzsch, H. Randel, T. Weyhermüller, A. A. Auer, C. Farès, C. Werlé, Chem Catalysis 2024, 4, 101078.
[8] A. Pierret, K. Magra, H. Lopez, T. Kauffmann, C. Denhez, I. Abdellah, C. Werlé, A. Vasseur, Chem. Commun. 2025, 61, 6296–6299.
