

Les interfaces jouent un rôle crucial dans un large éventail de domaines scientifiques et technologiques tels que l’adsorption, la catalyse et la synthèse de matériaux nanostructurés. En effet, les interfaces orientent drastiquement les processus d’adsorption et de sélectivité vis-à-vis d’un mélange de molécules. La modulation des propriétés physico-chimiques des interfaces permet de génèrer des sites actifs spécifiques influençant directement la cinétique et l’efficacité de l’adsorption. D’autre part, le rendement des réactions catalytiques peut être considérablement amélioré en optimisant les propriétés texturales du support, telles que la surface spécifique, la morphologie et la nature de sa structure électronique. Le contrôle de ces critères est fondamental pour créer de nouveaux matériaux aux propriétés interfaciales uniques et spécifiques aux applications visées.
En combinant notre expertise dans les domaines de la physico-chimie, la catalyse hétérogène et l’interprétation des processus d’adsorption, l’équipe InterACT (Interfaces, Adsorption et CaTalyse) travail à développer des solutions durables et efficaces pour relever les défis sociétaux actuels.
Plus concrètement, l’équipe InterACT mène des activités de recherche dans trois domaines principaux fortement liés entre eux :
- Réactivité chimique aux interfaces : Étude des phénomènes chimiques se produisant à l’interface entre différentes phases (solide/liquide, liquide/gaz, etc.), avec un intérêt particulier pour les mécanismes réactionnels, la cinétique et la réactivité.
- Ingénierie moléculaire à l’interface : Conception et synthèse de matériaux fonctionnelles pour contrôler et optimiser les propriétés des interfaces, avec des applications dans des domaines variés tels que la catalyse, les procèdes photo-actifs et la biomédecine.
- Catalyses pour un futur durable : Développement de catalyseurs innovants et des procédés catalytiques/chimiques/hybrides plus durables, visant à réduire l’impact environnemental et à valoriser les ressources renouvelables dont le CO2.
Fait marquant
L’équipe InterACT en Espagne pour lancer le nouveau projet de recherche « +C2FUe-LS »
Le projet de recherche « +C2FUe-LS » (Transformation du carbone recyclé en carburants pour tous les secteurs des transports par l’association d’un plasma sélectif et de la bio/chimiocatalyse dans des conditions douces), financé par le programme Horizon UE, vise à développer une technologie de rupture pour la production de carburants synthétiques renouvelables destinés aux transports, grâce à des procédés à haute efficacité énergétique.

Lancé la semaine dernière à Huesca et Saragosse (Espagne), le projet réunira sept partenaires issus de quatre pays de l’UE, sous la coordination de la Fondation Aragon pour l’Hydrogène (FHa). Il se concentrera sur la conversion du CO2 en alcools et en composés clés, notamment des carburants et des produits chimiques à haute valeur ajoutée, par la technologie plasma dans des conditions douces, à une température inférieure à 100 °C (plasma froid). Le réacteur final, alimenté exclusivement par de l’électricité renouvelable, du CO2 et de l’hydrogène vert, pourra produire sélectivement :
- des aldéhydes et des alcools légers pour les piles à combustible et une utilisation directe dans le transport maritime ;
- des composants essentiels pour l’industrie chimique ;
- des alcools de qualité supérieure pour les carburants d’aviation durables (SAF).
Grâce à ces recherches, +C2FUe-LS contribuera à la réalisation de l’objectif ambitieux de l’Union européenne : une économie moins dépendante des énergies fossiles.

Publication
Jérôme Rey, Céline Chizallet, Dario Rocca, Tomas Bučko, Michael Badawi*, Reference-Quality Free Energy Barriers in Catalysis from Machine Learning Thermodynamic Perturbation Theory, Angewandte Chemie International Edition, 63 (2024) e202312392 (Impact Factor = 16,9). https://doi.org/10.1002/anie.202312392
Pour la toute première fois, nous avons réussi à calculer des énergies libres d’activation de réactions catalytiques dans un matériau poreux (zéolithe) au plus haut niveau de théorie disponible pour les solides (approximation de la phase aléatoire – RPA) et à température finie, en combinant des simulations de dynamique moléculaire ab initio avec des méthodes d’intelligence artificielle. Nous démontrons que l’utilisation du haut niveau de théorie RPA est nécessaire pour combler l’écart entre les valeurs expérimentales et calculées. Ces transformations, catalysées par les zéolites et les intermédiaires cationiques et les états de transition, sont les éléments constitutifs de nombreuses transformations chimiques pour la valorisation de paraffines à longue chaîne provenant, par exemple, de déchets plastiques, d’huiles végétales, ou de pétroles bruts. Par rapport aux barrières d’énergie libre calculées au niveau de production PBE + D2 de la théorie via une dynamique moléculaire ab initio contrainte, les barrières calculées au niveau RPA par l’application de la théorie des perturbations thermodynamiques d’apprentissage automatique (MLPT) montrent une diminution significative de la réaction d’isomérisation et une augmentation d’une ampleur similaire pour la réaction de craquage, donnant un accord sans précédent avec les résultats obtenus par les expériences et la modélisation cinétique.
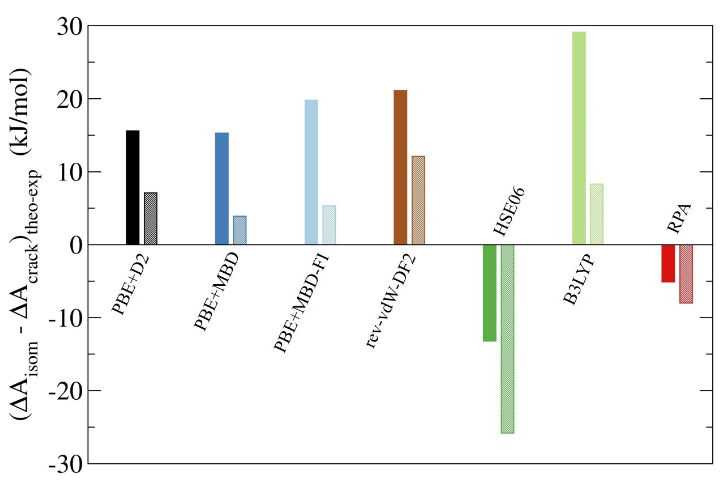
Écart par rapport à l’expérience de la différence des énergies libres d’activation aux différents niveaux de théorie étudiés entre les réactions d’isomérisation et de craquage (barres sombres : isomérisation entre les cations tertiaires diramifiés et tribranchés ; barres claires : isomérisation entre les cations tribranchés tertiaires à dibranchés).
